

ライソゾーム病の酵素製剤
ライソゾーム病の全体的な診断・治療についての現状、東京エリアにおける新生児マススクリーニングの状況についてご説明いたします。
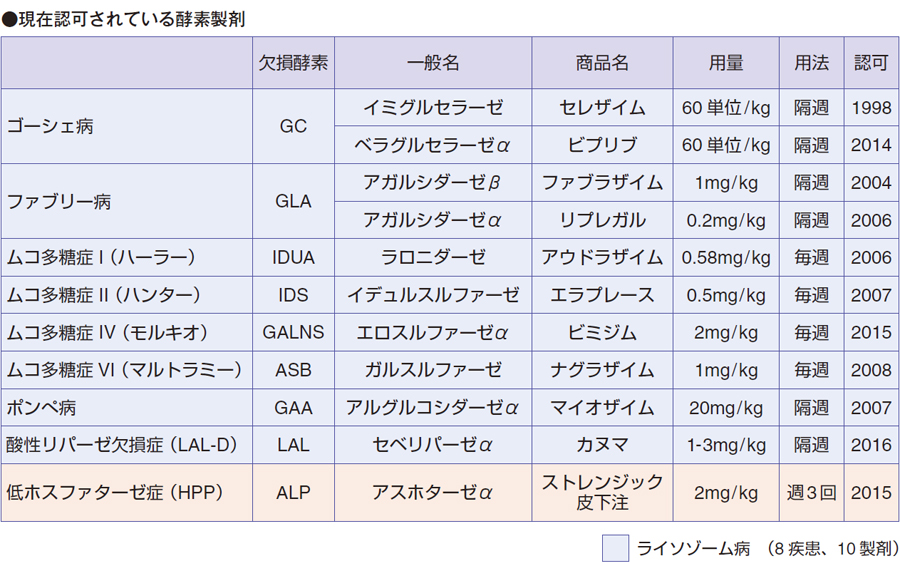
現在、ライソゾーム病に認可されている酵素製剤は、ゴーシェ病は2 剤、ファブリー病は2 剤、ムコ多糖症(MPS)Ⅰ、Ⅱ、Ⅳ、Ⅵ、ポンペ病、酸性リパーセ欠損症(LAL-D)、は各1 製剤。8 疾患に対し10 製剤が使用されています。ポンペ病に対するマイオザイムの用量が非常に突出していますが、これが原因で、Infusion associated reaction(IAR)などのアレルギー反応が多いと考えられます。マイオザイム投与による抗体発現が多いことは、深尾先生がご指摘のとおり、問題の1つだと考えています。
現在、ライソゾーム病に認可されている酵素製剤は、ゴーシェ病は2 剤、ファブリー病は2 剤、ムコ多糖症(MPS)Ⅰ、Ⅱ、Ⅳ、Ⅵ、ポンペ病、酸性リパーセ欠損症(LAL-D)、は各1 製剤。8 疾患に対し10 製剤が使用されています。ポンペ病に対するマイオザイムの用量が非常に突出していますが、これが原因で、Infusion associated reaction(IAR)などのアレルギー反応が多いと考えられます。マイオザイム投与による抗体発現が多いことは、深尾先生がご指摘のとおり、問題の1つだと考えています。
ファブリー病患者の自然経過
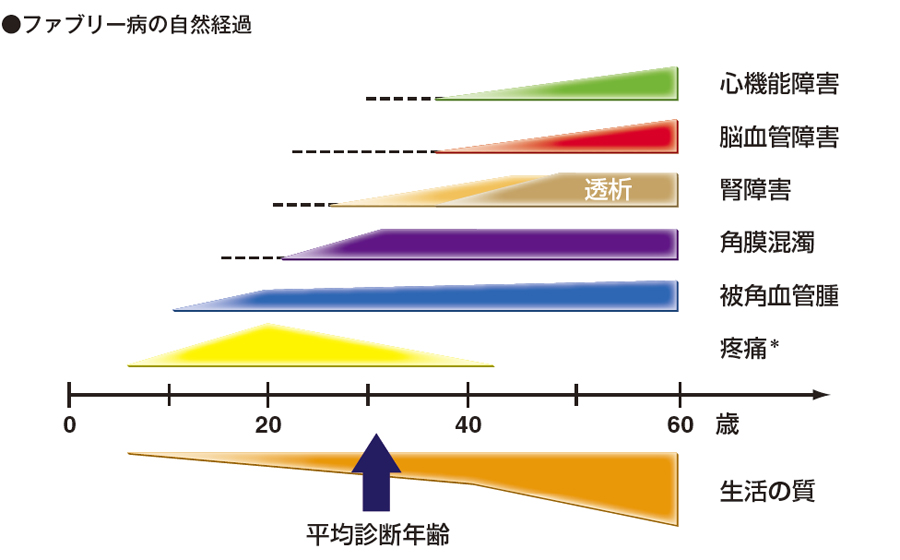
ファブリー病の自然経過についてご説明します。最初、四肢の疼痛が起きてきます。腹部とかベルトのあたりに診られることが多い被角血管腫が発現する人もいますし、まったく発現しない人もいます。スポーク状の角膜混濁は眼科に行かないと診断されませんが、角膜混濁は女性のファブリー病を見つけるのに結構よいポイントになっています。
ファブリー病では腎障害が大きな問題です。治療をしないと、40 歳頃には全例透析になってしまいます。これを未然に防ぐことが酵素補充療法(ERT)の主要目的の1つです。
ファブリー病では、脳梗塞などの脳血管障害が起こりやすくなります。知的障害は起こらないとされています。
それから、心機能障害が腎障害よりも問題になっています。心筋肥大、心筋症、致死的不整脈は本当に怖く、心臓合併症が一番怖いのではないかなと思っています。女性はむしろ心臓に障害が起きやすいので、非常に気をつけなくてはいけないと考えています。
自然経過で最初に出現する四肢疼痛は、特に小児の患者さんにとっては一番つらい症状で、疼痛発症の9 割は4 歳~ 12 歳、主に小学生の頃で、非常につらい。ガバペンチンとテグレトールが第一選択薬ですが、全く効かない人も多いようで、かなり深刻です。患者さん本人にとっては、初期の症状では四肢疼痛が一番つらいようです。
平均診断年齢ですが、2017 年のJournal of Nephropathology では、オーストラリアが28.6 歳、2016 年のJournal of Human Genetetics では、中国が38.5 歳。日本は2015 年の日本小児科学会雑誌で28.3 歳でした。この付近の年齢では疼痛、被角血管腫だけでなく、角膜混濁を疑わないと、小児科ではみつからない状況なので、角膜混濁を疑って眼科に検査を依頼するというスキルが必要になってきます。腎障害、タンパク尿が出てからの診断では遅いわけです。
ファブリー病では腎障害が大きな問題です。治療をしないと、40 歳頃には全例透析になってしまいます。これを未然に防ぐことが酵素補充療法(ERT)の主要目的の1つです。
ファブリー病では、脳梗塞などの脳血管障害が起こりやすくなります。知的障害は起こらないとされています。
それから、心機能障害が腎障害よりも問題になっています。心筋肥大、心筋症、致死的不整脈は本当に怖く、心臓合併症が一番怖いのではないかなと思っています。女性はむしろ心臓に障害が起きやすいので、非常に気をつけなくてはいけないと考えています。
自然経過で最初に出現する四肢疼痛は、特に小児の患者さんにとっては一番つらい症状で、疼痛発症の9 割は4 歳~ 12 歳、主に小学生の頃で、非常につらい。ガバペンチンとテグレトールが第一選択薬ですが、全く効かない人も多いようで、かなり深刻です。患者さん本人にとっては、初期の症状では四肢疼痛が一番つらいようです。
平均診断年齢ですが、2017 年のJournal of Nephropathology では、オーストラリアが28.6 歳、2016 年のJournal of Human Genetetics では、中国が38.5 歳。日本は2015 年の日本小児科学会雑誌で28.3 歳でした。この付近の年齢では疼痛、被角血管腫だけでなく、角膜混濁を疑わないと、小児科ではみつからない状況なので、角膜混濁を疑って眼科に検査を依頼するというスキルが必要になってきます。腎障害、タンパク尿が出てからの診断では遅いわけです。

ファブリー病の発症から診断までの時間の遅れ
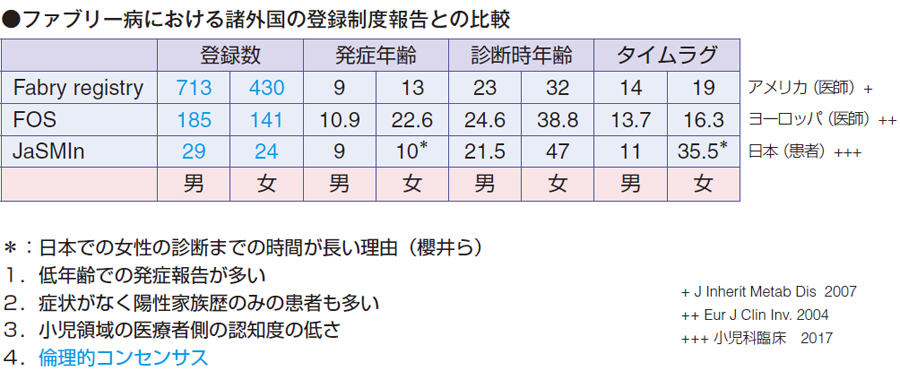
慈恵医大の櫻井謙先生がファブリー病における諸外国の登録制度報告の比較を小児科臨床に投稿しています。ファブリー病にはかなり大規模なレジストリーがあり、1 つはアメリカ中心のFabry registry です。登録数は男性713 人、女性430 人で、発症年齢は男性9 歳、女性13 歳、診断時年齢は男性23 歳、女性32 歳で、タイムラグは男性14 年、女性19 年でした。
もう1 つはヨーロッパ中心のFOS(Fabry Outcome Survey)で、登録数は男性185 人、女性141 人で、発症年齢は男性10 歳、女性22 歳という大体思春期が終わったころで、診断時は男性24 歳、女性38 歳、タイムラグはアメリカの方が女性は少し遅い程度です。
日本はJaSMIn という患者登録制度があり、男性29 人、女性24 人で、発症年齢は男性9 歳、女性10 歳となっています。診断時年齢は男性21 歳、女性47 歳と女性でかなり遅れています。タイムラグが男性11 年、女性35.5 年と、アメリカ、ヨーロッパに比べて一番長くなっています。Fabry registry、FOS は医師が報告するレジストリーであるのに対し、JaSMIn は患者さんから報告する登録です。登録数もだいぶ少なくなっています。
櫻井先生は日本での女性の診断までの時間が長い理由について、1 つは低年齢の発症報告が多いこと、発症の症状として、JaSMIn に陽性家族歴のみの患者(症状がなくても発症ということになっている患者)が多いこと、もう1 つは、小児領域の小児科医の認知度が低いことを挙げています。
あと、私は倫理的なコンセンサスも原因だと考えています。特に女性の患者さんは、思春期以降で発症することが多く、あまり早く見つけて長い無症状期間があると、そんなに早く見つけてしまっていいのか、治療法はあるなら治療はあとでもいいといった、側面があると推測します。
ファブリー病の発症から診断までの遅れの期間は、FOS の結果をみると、2001~ 2006 年に比べ、2007~ 2013 年で遅れの期間は短くなっています。
一方、ファブリー病は診断後、いつから治療するかが問題になっています。今のコンセンサスとしては、男児は小児期・学童期前後で、女児は思春期前後と考えられています。診断から治療までの期間も短縮しているというのが世界的な趨勢のようです。
もう1 つはヨーロッパ中心のFOS(Fabry Outcome Survey)で、登録数は男性185 人、女性141 人で、発症年齢は男性10 歳、女性22 歳という大体思春期が終わったころで、診断時は男性24 歳、女性38 歳、タイムラグはアメリカの方が女性は少し遅い程度です。
日本はJaSMIn という患者登録制度があり、男性29 人、女性24 人で、発症年齢は男性9 歳、女性10 歳となっています。診断時年齢は男性21 歳、女性47 歳と女性でかなり遅れています。タイムラグが男性11 年、女性35.5 年と、アメリカ、ヨーロッパに比べて一番長くなっています。Fabry registry、FOS は医師が報告するレジストリーであるのに対し、JaSMIn は患者さんから報告する登録です。登録数もだいぶ少なくなっています。
櫻井先生は日本での女性の診断までの時間が長い理由について、1 つは低年齢の発症報告が多いこと、発症の症状として、JaSMIn に陽性家族歴のみの患者(症状がなくても発症ということになっている患者)が多いこと、もう1 つは、小児領域の小児科医の認知度が低いことを挙げています。
あと、私は倫理的なコンセンサスも原因だと考えています。特に女性の患者さんは、思春期以降で発症することが多く、あまり早く見つけて長い無症状期間があると、そんなに早く見つけてしまっていいのか、治療法はあるなら治療はあとでもいいといった、側面があると推測します。
ファブリー病の発症から診断までの遅れの期間は、FOS の結果をみると、2001~ 2006 年に比べ、2007~ 2013 年で遅れの期間は短くなっています。
一方、ファブリー病は診断後、いつから治療するかが問題になっています。今のコンセンサスとしては、男児は小児期・学童期前後で、女児は思春期前後と考えられています。診断から治療までの期間も短縮しているというのが世界的な趨勢のようです。
酵素補充療法の10 年目の評価
ファブリー病に対する酵素補充療法(ERT)は承認されて約10 年が経過しました。その効果に対する評価が多数報告されています。Anderson らは289 名の追跡研究を報告していますが、それによると、経時的に有意な左室心筋重量の低下、女性ではeGFR(糸球体ろ過量)の上昇が確認されています。一方、痛み、聴力、TIA(脳虚血発作)のイベントには有意差のある効果はみられませんでした。
聴力は効いているというデータもみられます。めまいと聴力の低下は非常に深刻な合併症です。
痛みに関してはガバペンチンとテグレトールなどの効果と酵素補充療法の効果に関連はなく、酵素補充療法だけでみると効果は乏しいようです。
聴力は効いているというデータもみられます。めまいと聴力の低下は非常に深刻な合併症です。
痛みに関してはガバペンチンとテグレトールなどの効果と酵素補充療法の効果に関連はなく、酵素補充療法だけでみると効果は乏しいようです。
ファブリー病に対するシャペロン療法
ファブリー病に対するシャペロン療法についてご説明します。シャペロン療法はまだ承認されていません。鈴木義之先生らは患者細胞で変異α- galactosidase A(α- GalA)がガラクトース存在下で安定化することを発見、更にガラクトース類似体の1-deoxy-galactonojirimycin(DGJ)がR301Q 変異のα- GalA を安定化し、酵素活性を8 倍に増加、グロボトリアオシルセラミド(Gb3)を減少させることを1999 年、Nature Medicine に報告しました。
Amicus 社がこのDGJ を経口薬Migalastat HCL としてグラクソ・スミスクライン(GSK)と共同で開発し、2012 年までに第三相試験を終了。感受性のある遺伝子型には有効性を示しています。
その後、53 名の患者さんにERT から変更投与し、腎機能、左室心筋重量、安全性、血清GB3 値にERT と同等の効果を示すことが判明しました。ERT と大体同等のレベルで効いてきているのではないかということで、恐らく2018 年の夏以降、治験が日本でも進むかもしれないということです。
もう1 つ、酵素補充療法とシャペロン療法の組み合わせ治療(酵素補充2 時間前に服用)が、第二相試験までで感受性のある遺伝子型患者には有効性を示しました。ただ、その後あまりうまく進んでいないようです。ファブリー病に関しても、シャペロン療法が2018 年以降でてくる予定なので、治療の選択肢が増えていくと考えられています。
Amicus 社がこのDGJ を経口薬Migalastat HCL としてグラクソ・スミスクライン(GSK)と共同で開発し、2012 年までに第三相試験を終了。感受性のある遺伝子型には有効性を示しています。
その後、53 名の患者さんにERT から変更投与し、腎機能、左室心筋重量、安全性、血清GB3 値にERT と同等の効果を示すことが判明しました。ERT と大体同等のレベルで効いてきているのではないかということで、恐らく2018 年の夏以降、治験が日本でも進むかもしれないということです。
もう1 つ、酵素補充療法とシャペロン療法の組み合わせ治療(酵素補充2 時間前に服用)が、第二相試験までで感受性のある遺伝子型患者には有効性を示しました。ただ、その後あまりうまく進んでいないようです。ファブリー病に関しても、シャペロン療法が2018 年以降でてくる予定なので、治療の選択肢が増えていくと考えられています。
ポンぺ病の発症から診断までの時間の遅れ
次に、ポンペ病について解説します。乳児型ポンぺ病に対する酵素補充療法(ERT)の効果を検討したKishnaniらの報告は衝撃的なもので、自然歴では15 カ月で全例亡くなってしまうのに対し、ERT では全例で生存していたという結果でしたが、データをよくみると、自然歴では4 カ月ぐらいから死亡例が出てきています。つまり、4 カ月齢以前に診断し、治療を開始する必要があるともいえるわけです。
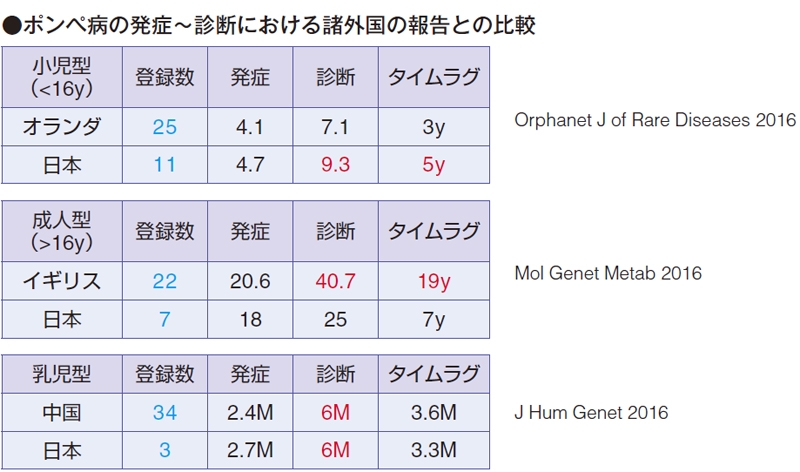
下図に最近の遅発型(小児型と成人型)と乳児型のポンぺ病の発症から診断までのタイムラグについてまとめてみました。
小児型の場合、オランダでは4 歳発症、7 歳診断で、タイムラグが3 年、日本では4 歳発症、9 歳診断で、タイムラグ5 年でした。オランダに少し遅れている状況でした。
成人型の場合、イギリス(一部南アフリカ)では20 歳発症、40 歳診断、タイムラグ19 年、日本では18 歳発症、25 歳診断で、タイムラグは7 年でした。成人型ではイギリスより早くなっています。
乳児型の場合、中国のデータを示しますが、2 カ月齢発症、6 カ月診断、タイムラグは3.6 カ月と我々のデータとほぼ同じです。ただ、4 カ月齢から死亡例が出ているので、6 カ月で診断するのはやや遅いかもしれません。診断時期を早める努力が必要です。
私のポンぺ病患者4 例に対するデータでも、酵素補充療法を早く開始した方がmanual muscle testing(MMT)や重症度スコア(WGs)がよいという結果でした。
下図に最近の遅発型(小児型と成人型)と乳児型のポンぺ病の発症から診断までのタイムラグについてまとめてみました。
小児型の場合、オランダでは4 歳発症、7 歳診断で、タイムラグが3 年、日本では4 歳発症、9 歳診断で、タイムラグ5 年でした。オランダに少し遅れている状況でした。
成人型の場合、イギリス(一部南アフリカ)では20 歳発症、40 歳診断、タイムラグ19 年、日本では18 歳発症、25 歳診断で、タイムラグは7 年でした。成人型ではイギリスより早くなっています。
乳児型の場合、中国のデータを示しますが、2 カ月齢発症、6 カ月診断、タイムラグは3.6 カ月と我々のデータとほぼ同じです。ただ、4 カ月齢から死亡例が出ているので、6 カ月で診断するのはやや遅いかもしれません。診断時期を早める努力が必要です。
私のポンぺ病患者4 例に対するデータでも、酵素補充療法を早く開始した方がmanual muscle testing(MMT)や重症度スコア(WGs)がよいという結果でした。
ポンぺ病に対する新しい治療法
ポンぺ病に対しては、いくつかの新しい治療法が模索されています。
最近、JCR ファーマが開発したJ-Brain Cargo® という日本初の血液脳関門通過技術を適用した酵素製剤について、MPSⅡ型だけでなくポンペ病でも治験が始まろうとしています。これは抗トランスフェリン受容体抗体を酵素に付けて、Blood Brain Barrier(BBB)を通過させようという技術です。骨格筋も通りやすくなるのではないかということです。
次世代マイオザイム(neoGAA)は今のマイオザイムをさらに進化させて、enhanced receptor biding and enzyme uptake ということで、Sanofi Genzyme が開発していて、2019 年以降に市場に入ってくるということでした。
他にもいろいろな会社の方法がありまして、Amicus はコスト的に厳しいかもしれないのですがシャペロンとの併用、Audentes はAAVウイルスベクターによる遺伝子治療を開発中です。
最近、JCR ファーマが開発したJ-Brain Cargo® という日本初の血液脳関門通過技術を適用した酵素製剤について、MPSⅡ型だけでなくポンペ病でも治験が始まろうとしています。これは抗トランスフェリン受容体抗体を酵素に付けて、Blood Brain Barrier(BBB)を通過させようという技術です。骨格筋も通りやすくなるのではないかということです。
次世代マイオザイム(neoGAA)は今のマイオザイムをさらに進化させて、enhanced receptor biding and enzyme uptake ということで、Sanofi Genzyme が開発していて、2019 年以降に市場に入ってくるということでした。
他にもいろいろな会社の方法がありまして、Amicus はコスト的に厳しいかもしれないのですがシャペロンとの併用、Audentes はAAVウイルスベクターによる遺伝子治療を開発中です。
ムコ多糖症の発症から診断までの時間の遅れ
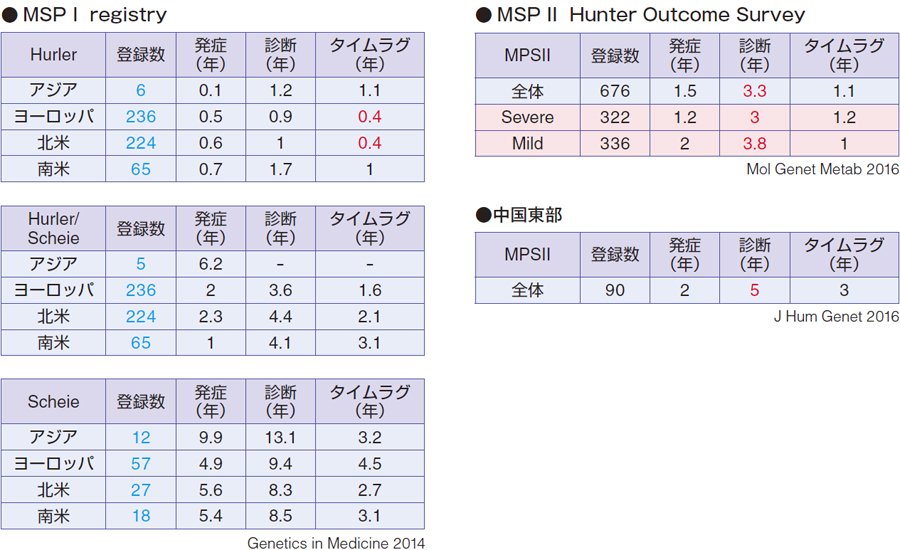
ムコ多糖症Ⅰ型では、MPS Ⅰ Registry という大きい登録システムがあり、アジア、ヨーロッパ、北米、南米に分けたデータがあります。アジアは登録数が6 例と少ないので比べられませんが、重症型のハーラーに関しては、ヨーロッパの発症は0.5 年(6 カ月ぐらい)、診断0.9 年(10 ~ 11 カ月ぐらい)で、タイムラグは0.4 年で診断はかなり早くなっています。ヨーロッパ・北米系はハーラーが圧倒的に多いということもあります。タイムラグも、アジア、南米より早いということです。ハーラー/シャイエあるいはシャイエになると、重症度が落ちてきますので、タイムラグ・診断時年齢も変わってきます。
ムコ多糖症Ⅱ型では、Hunter Outcome Survey(HOS)があり、登録全体数676 例で、発症1.5 歳、診断3.3 歳、タイムラグが1.1 年です。精神発達遅滞を伴うSevere とMild に分けていて、Severe では発症1.2 歳、診断3 歳、Mild では発症2 歳、診断3.8 歳で、タイムラグはSevere のほうがやや大きくなっています。
中国では90 例の登録の報告があって、発症2 歳、診断5 歳、タイムラグ3 年という結果です。欧米系のHOSよりタイムラグがやや大きい数字になっています。
ムコ多糖症Ⅱ型では、Hunter Outcome Survey(HOS)があり、登録全体数676 例で、発症1.5 歳、診断3.3 歳、タイムラグが1.1 年です。精神発達遅滞を伴うSevere とMild に分けていて、Severe では発症1.2 歳、診断3 歳、Mild では発症2 歳、診断3.8 歳で、タイムラグはSevere のほうがやや大きくなっています。
中国では90 例の登録の報告があって、発症2 歳、診断5 歳、タイムラグ3 年という結果です。欧米系のHOSよりタイムラグがやや大きい数字になっています。
造血幹細胞移植が標準治療適応となる先天代謝異常症
加藤班が作成した先天代謝異常症に対する造血幹細胞移植ガイドラインの中で、標準適応となるのはムコ多糖症Ⅰ型(重症型のハーラー)(年齢< 2 歳、DQ > 70)、Ⅱ型(ハンター)(重症型TypeD の早期)、副腎白質ジストロフィー(ALD)(Loes score < 9,PIQ > 80)、クラッベ病(GLD)(乳児型)の4 つです。日本はこの4 つをガイドラインとして標準治療適応に挙げています。標準適応というのはファーストチョイスとして考えてもいいよという意味合いです。
実はムコ多糖症Ⅱ型は、加藤班で新たに提唱していこうという日本の案です。欧米では効果がないということが一般的になっていますが、日本では症例数も多いし、ムコ多糖症の半分はⅡ型だということで、Ⅱ型も一応効果があるということを発信していこうということになったわけです。ただ、年齢としては2 歳未満で考えていきたいということにしています。
実はムコ多糖症Ⅱ型は、加藤班で新たに提唱していこうという日本の案です。欧米では効果がないということが一般的になっていますが、日本では症例数も多いし、ムコ多糖症の半分はⅡ型だということで、Ⅱ型も一応効果があるということを発信していこうということになったわけです。ただ、年齢としては2 歳未満で考えていきたいということにしています。

副腎白質ジストロフィー(ALD)と異染性白質ジストロフィー(MLD)に対する遺伝子治療
ここで遺伝子治療の話題を提供したいと思います。今、先天性代謝異常症で遺伝子治療がうまくいっているのは副腎白質ジストロフィー(ALD)と異染性白質ジストロフィー( metachromatic leukodystrophy:MLD)の2つです。ex vivo でレンチウイルス(LV)に欠損遺伝子を組み込んで自家骨髄移植をやる方法は、一番うまくいっている遺伝子治療です。ALD もMLD も骨髄移植と同等の結果になっており、ALD では進行抑制、MLD では発症予防に成功しています。現時点では脱髄の進行を強力に抑制することができますが、完成した脱髄を戻すことは遺伝子治療でも難しいようです。
これらの疾患に対しても早期発見が重要であり、臨床症状、家族歴から疑う力が必要で、スクリーニングをどう組み込んでいくかが、今後、議論になってくると思います。
これらの疾患に対しても早期発見が重要であり、臨床症状、家族歴から疑う力が必要で、スクリーニングをどう組み込んでいくかが、今後、議論になってくると思います。
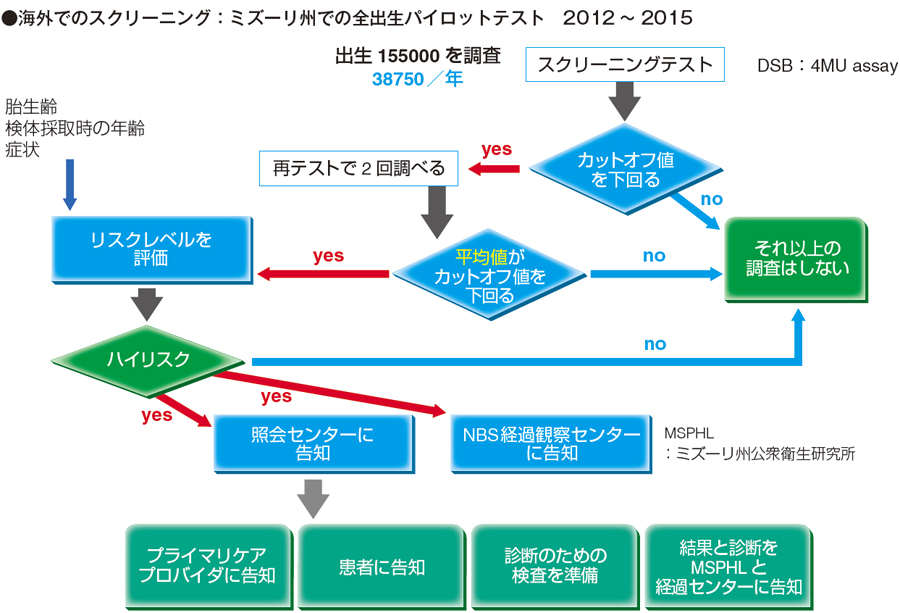
ミズーリ州で実施された新生児スクリーニング
下図は、成田先生のお話の中にもあったミズーリ州の新生児スクリーニングのパイロットテストのフローチャートです。
ろ紙血はタンデムマスではなく4MU で酵素活性を測定して、カットオフ値を下回るか下回らないかでいろいろな体制を敷いています。行政が絡んでいるので、それなりに予算もついていると思われます。ゴーシェ病に関しても、従来の報告と同じような疾患頻度であることが改めて判明しています。
ただ、ポンぺ病などでpseudodeficiency がある場合は、コストの問題はありますが、タンデムマスを使用することでしっかり区別できます。4MU で最初にやるのは決して悪い方法ではないと思いますが、このあたりは今後の課題といえます。
ろ紙血はタンデムマスではなく4MU で酵素活性を測定して、カットオフ値を下回るか下回らないかでいろいろな体制を敷いています。行政が絡んでいるので、それなりに予算もついていると思われます。ゴーシェ病に関しても、従来の報告と同じような疾患頻度であることが改めて判明しています。
ただ、ポンぺ病などでpseudodeficiency がある場合は、コストの問題はありますが、タンデムマスを使用することでしっかり区別できます。4MU で最初にやるのは決して悪い方法ではないと思いますが、このあたりは今後の課題といえます。
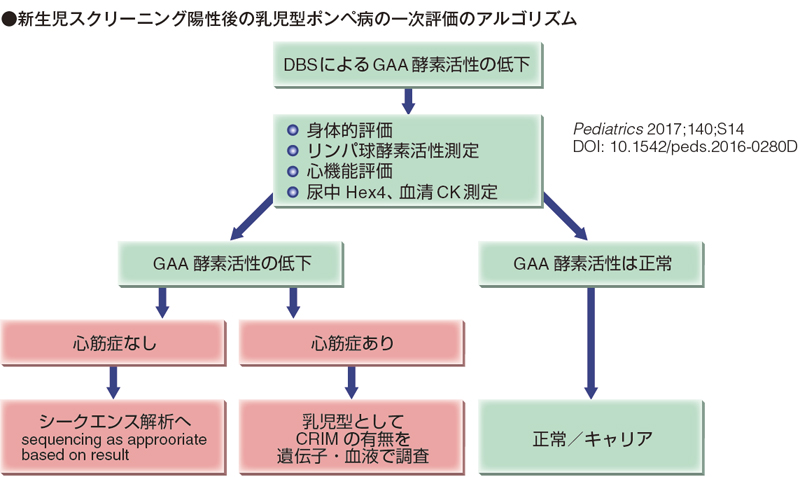
次図は奧山先生が2017 年、Pediatrics に掲載した新生児スクリーニング陽性後の乳児型ポンペ病の一次評価のアルゴリズムです。最初はacid α-glucosidase (GAA)が低い活性で、シークエンスを絡めない状況で、心筋症のあるなしで分けていくというシンプルなやり方です。心筋症があったら乳児型。CRIM の有無をチェックして、detect していく。
ただ、最近は次世代シークエンサー(NGS)も出てきて、変異を最初から絡めていって、2 つの病原性変異があったら一応classical かlate onset のどちらかですが、これも心筋症のあるなしで分けていく。1 つしかなくて、もう1 つは擬陽性(pseudodeficiency)や遺伝子多型(Polymorphism)だったら、血液をはかり直して見ていく。ボーダーラインだったら、pathogenic variant のキャリアかpseudodeficiency なのか。病原性変異がなかったらもう一回やり直して、やっぱり欠損があったら戻るというアルゴリズムがしっかり推奨されてきているようです。
ただ、最近は次世代シークエンサー(NGS)も出てきて、変異を最初から絡めていって、2 つの病原性変異があったら一応classical かlate onset のどちらかですが、これも心筋症のあるなしで分けていく。1 つしかなくて、もう1 つは擬陽性(pseudodeficiency)や遺伝子多型(Polymorphism)だったら、血液をはかり直して見ていく。ボーダーラインだったら、pathogenic variant のキャリアかpseudodeficiency なのか。病原性変異がなかったらもう一回やり直して、やっぱり欠損があったら戻るというアルゴリズムがしっかり推奨されてきているようです。
東京地区の新生児スクリーニングの現状
最後に遠藤先生からの宿題ですが、東京都予防医学協会の石毛先生からヒアリングした東京地区の新生児スクリーニングの現状についてお話します。東京は実は非常に遅れています。実施可能な測定項目あるいは現状は下記の通りです。ムコ多糖症に関しては、大阪市立大学で共同研究をされていましたが、中止になっている状況です。
●ファブリー病 尿GL3 TMS、尿・血漿GLA(DBS)4MU microplate
●ポンぺ病 血漿GAA(DBS)4MU microplate
●MPS Ⅰ / Ⅱ 血漿IDUA/IDS(DBS)TMS (現在中止)
●ゴーシェ病 未施行
東京都は年間出生が10 万人です。以前、ファブリー病で試験的にスクリーニングを実施した経験があり、パンフレットと同意書を作成し、病院を全部やるのは無理なので、倫理委員会を通過した日大や慈恵などの病院を特定して、1 万人ほどの規模でスクリーニングを実施している状況です。
今後、ミズーリ州のようにライソゾーム病をセットで始める場合、同意書・倫理委員会対策を行い、タンデムマスあるいは4MU microplate reader などの専用機器を用意することが必要になるとの見解を石毛先生から頂戴しました。
●ファブリー病 尿GL3 TMS、尿・血漿GLA(DBS)4MU microplate
●ポンぺ病 血漿GAA(DBS)4MU microplate
●MPS Ⅰ / Ⅱ 血漿IDUA/IDS(DBS)TMS (現在中止)
●ゴーシェ病 未施行
東京都は年間出生が10 万人です。以前、ファブリー病で試験的にスクリーニングを実施した経験があり、パンフレットと同意書を作成し、病院を全部やるのは無理なので、倫理委員会を通過した日大や慈恵などの病院を特定して、1 万人ほどの規模でスクリーニングを実施している状況です。
今後、ミズーリ州のようにライソゾーム病をセットで始める場合、同意書・倫理委員会対策を行い、タンデムマスあるいは4MU microplate reader などの専用機器を用意することが必要になるとの見解を石毛先生から頂戴しました。

まとめ
以上の内容を下記にまとめて列記します。
●ファブリー病
日本での女性の診断の遅れ:思春期以降の発症なので乳児期スクリーニングに対する批判もあり。男性は諸外国に遅れは取っていないが、診断はより早くする必要がある(世界の趨勢として、診断も治療導入も早くなっている)。
●ゴーシェ病
Ⅱ型を見つけることに議論も。近年、中枢移行する酵素製剤、基質合成阻害剤が開発中。
●ポンぺ病
遅発型のうち小児型でオランダよりやや発症から診断までのタイムラグが大きい。乳児・遅発型ともスクリーニングによる早期発見のメリットが大きい。
●ムコ多糖症Ⅰ・Ⅱ型
造血幹細胞移植の適応は2 歳未満。中枢移行製剤も治験へ。
1 歳半までの早期発見が課題(世界的にはMPS IH で1 歳、MPS II で3 歳で診断されている)。
● ALD やMLD
治験が進むALD・MLD の遺伝子治療も発見が遅れると効果が限定されるため、スクリーニングが重要。
●新生児スクリーニング
メリット:早期治療導入への備えと、正確な疾患頻度の把握
課題:
1. LC/MSMS などの使用機器は専用のものを購入・導入する必要あり
2. 同意取得・倫理委員会の対策(同意書・パンフレット作製)、手間とコスト
3. 倫理的コンセンサス
4. 実用・資金面では自治体とのリンクがないと厳しい
![]()